Clinical features
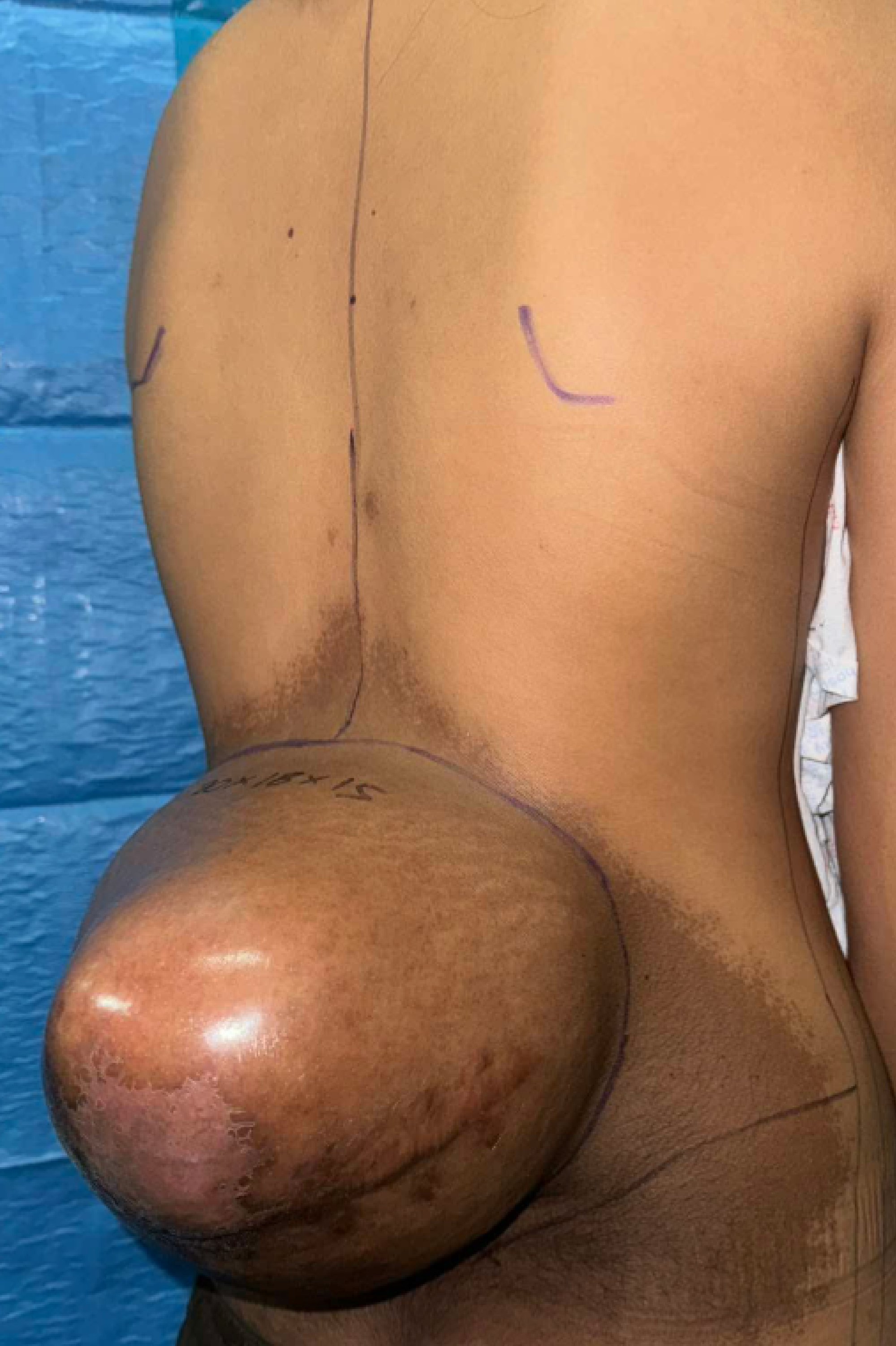
At birth, the patient was noted to have a hyperpigmented patch over the lower back. Approximately three years prior to the current admission, a mass measuring about 1 cm developed at the same site and gradually increased in size. Four months before admission, the mass rapidly enlarged to approximately 7 cm, prompting medical consultation. An initial ultrasound demonstrated a fluid-filled vascular mass, and subsequent biopsy revealed a spindle cell neoplasm. One month prior to admission, the patient consulted a private spine surgeon who obtained magnetic resonance imaging (MRI) of the thoracolumbar spine, which showed an encapsulated, skin-covered, complex cystic soft tissue mass in the posterior lumbar region. Based on the MRI findings and biopsy results, the patient was initially diagnosed with a spindle cell neoplasm and was advised to undergo immediate surgical intervention. Due to financial constraints, she was referred to the Orthopedic Service of our institution, where a wide resection of the lumbar mass with soft tissue reconstruction was planned. The patient denied lower extremity weakness or paresthesia. There was no family history of similar hyperpigmented lesions or masses, and she had no history of radiation exposure.On physical examination, a well-demarcated cystic mass measuring approximately 14 × 14 × 8 cm was observed over the lower back (Figure 1). The mass was soft, non-tender, and non-erythematous, with no evidence of discharge or necrosis, though a transverse incision scar from a prior biopsy was present. An overlying hyperpigmented patch with irregular borders extended from the thoracolumbar region to the superior gluteal area and contained sparse superficial hair. No café-au-lait macules were identified. The rest of the physical examination findings were unremarkable. Based on the patient’s clinical history and examination findings, a spindle cell neoplasm was initially suspected.
Diagnostics and therapeutics
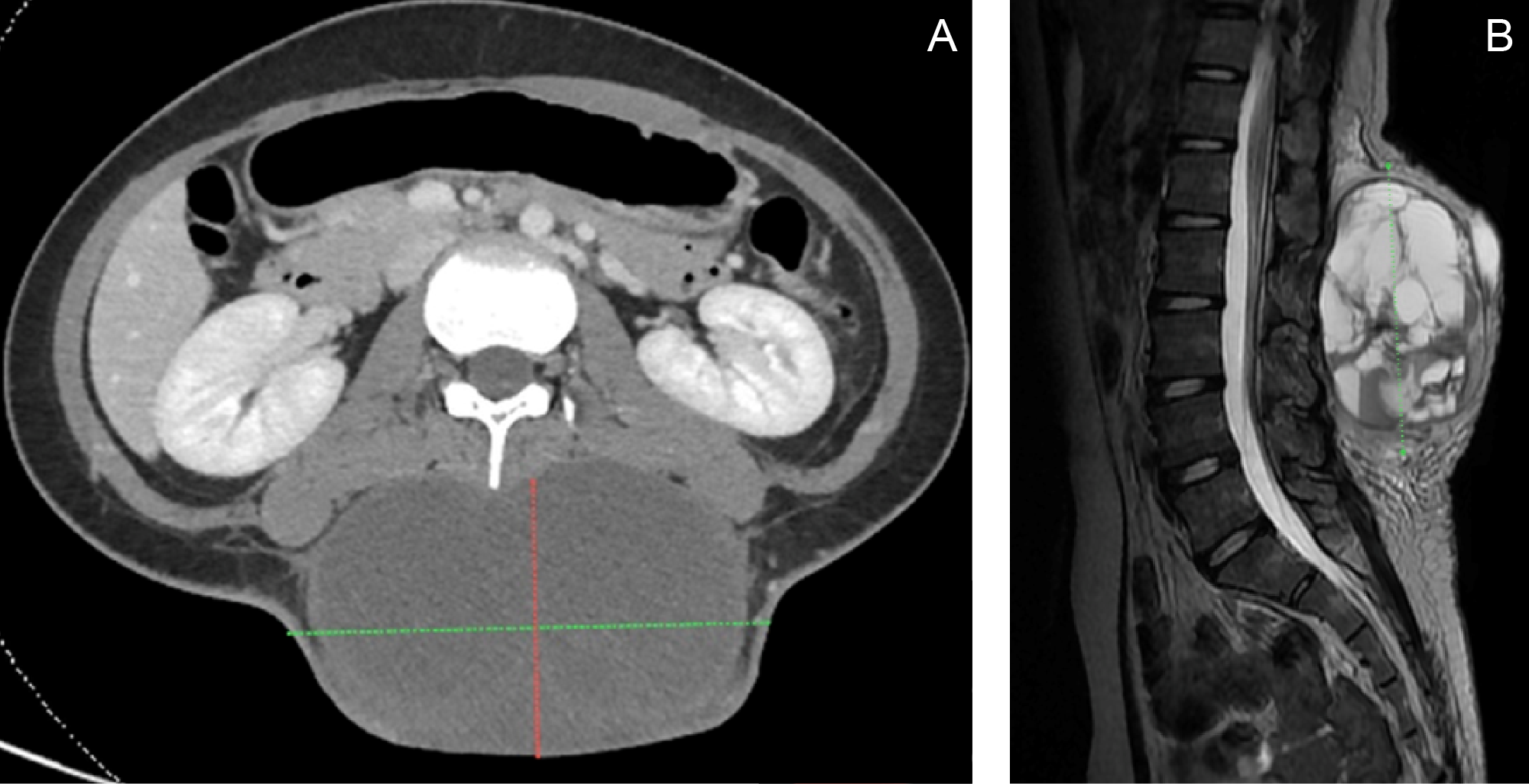
A CT scan and MRI prior to admission revealed an encapsulated, skin-covered, complex cystic soft tissue mass measuring 11.14 × 4.2 × 12.03 cm in the posterior lumbar region, extending from L1 to L5, without evidence of spinal extension (Figure 2). The imaging characteristics demonstrated overlapping features with plexiform neurofibromas, contributing to diagnostic uncertainty.The patient was initially admitted under the Orthopedic Service. On admission, chest and lumbosacral spine imaging confirmed the posterior lumbar mass and additionally revealed intervertebral disc disease at L2–L4, with possible anterolisthesis at L2–L3. She was subsequently referred to Pediatric Oncology and Dermatology for multidisciplinary comanagement. Dermatologic evaluation identified a congenital melanocytic nevus corresponding to the hyperpigmented patch present since birth. The patient was also referred to Plastic and Reconstructive Surgery for planning of soft tissue reconstruction.
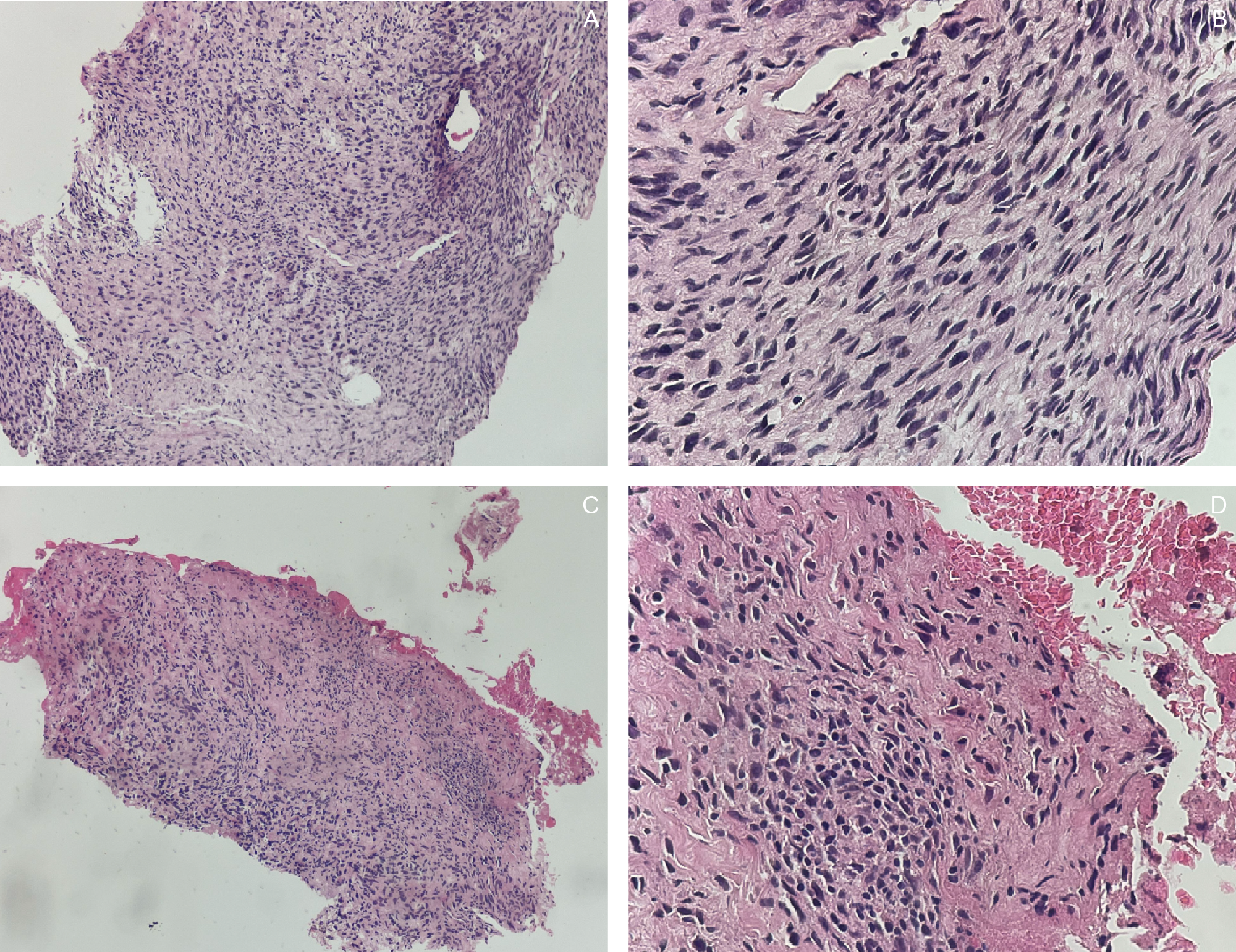
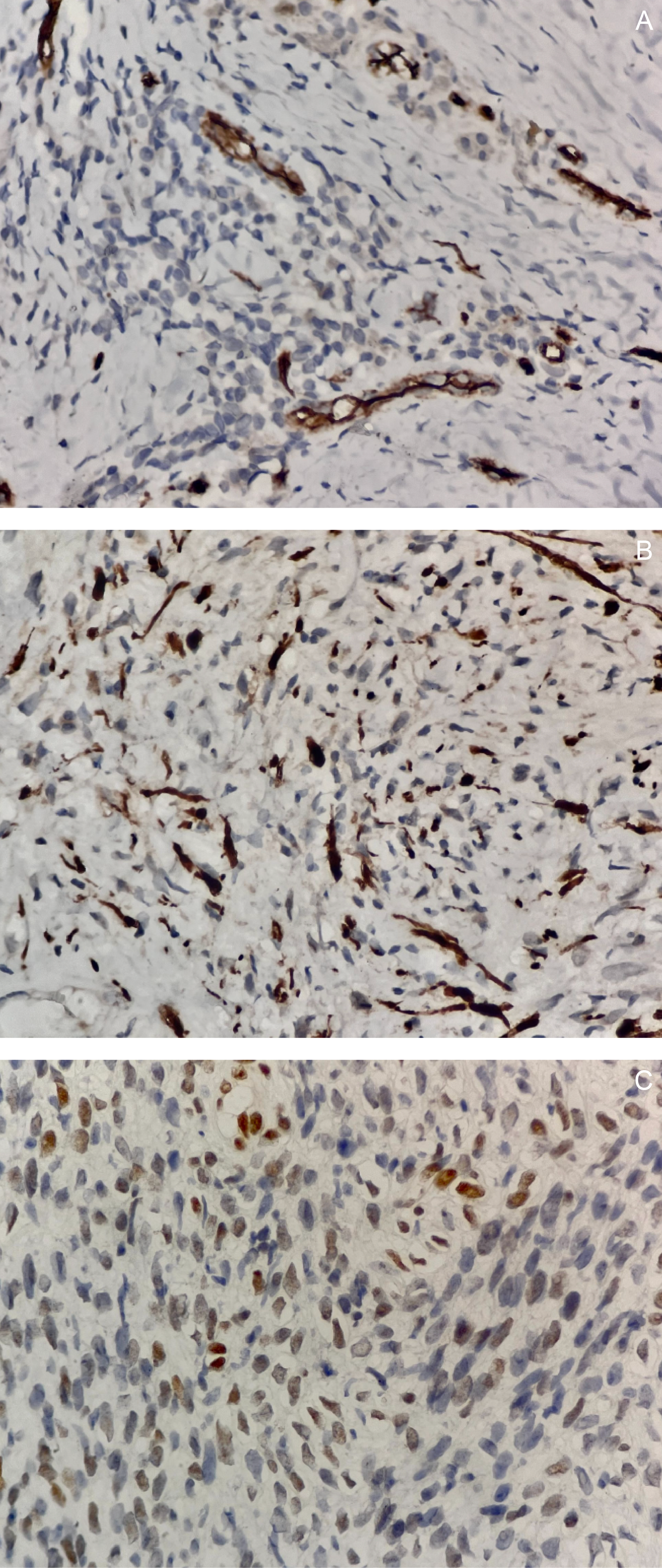
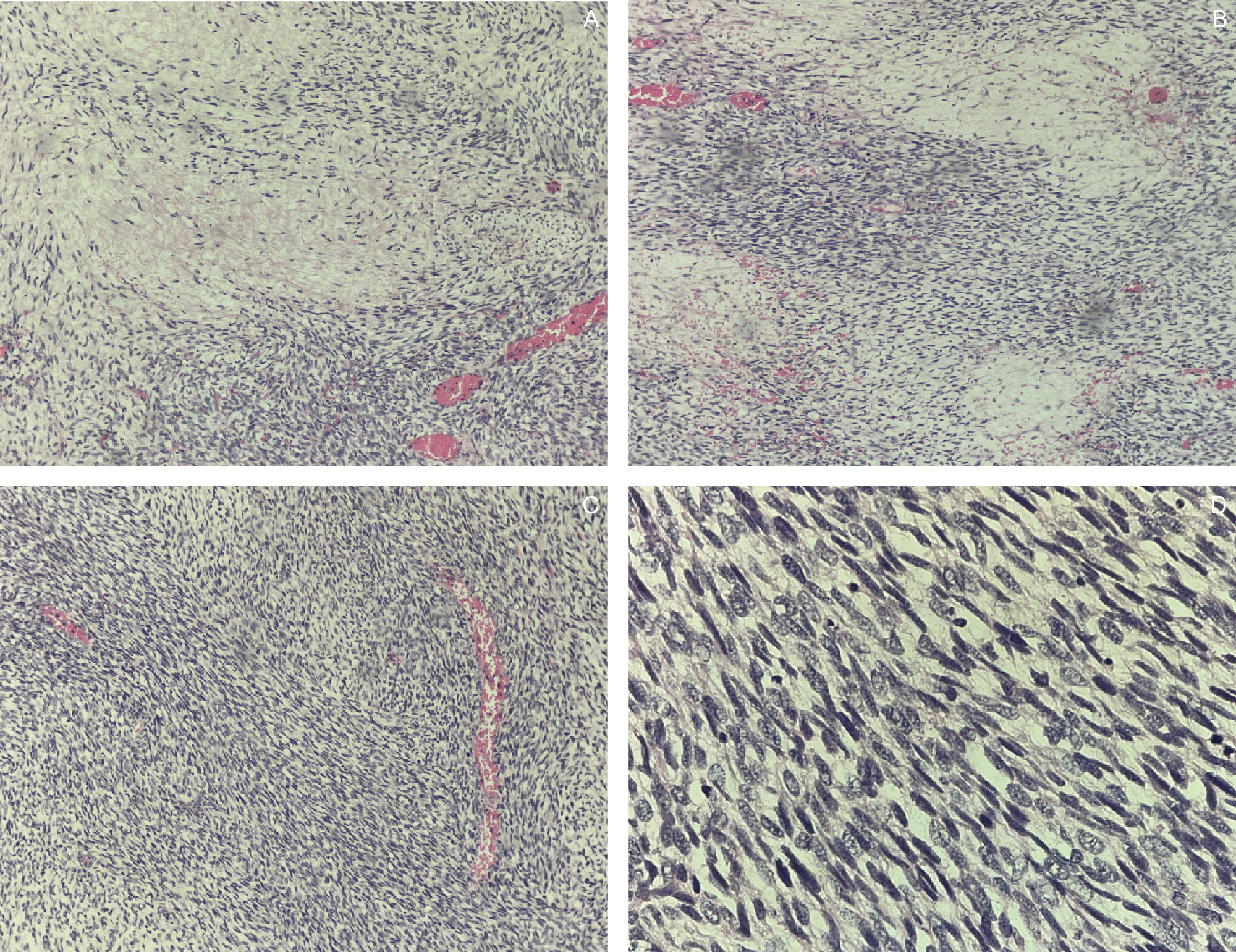
An ultrasound-guided core needle biopsy with frozen section demonstrated spindle cells with bland nuclear features, indistinct cytoplasm, inconspicuous nucleoli, focal necrosis, and rare mitotic figures. Histopathologic findings were consistent with a spindle cell sarcoma of the lower back (Figure 3). Immunohistochemical (IHC) staining showed positivity for S100, TLE-1, and CD34, supporting the diagnosis of a nerve sheath tumor (Figure 4).
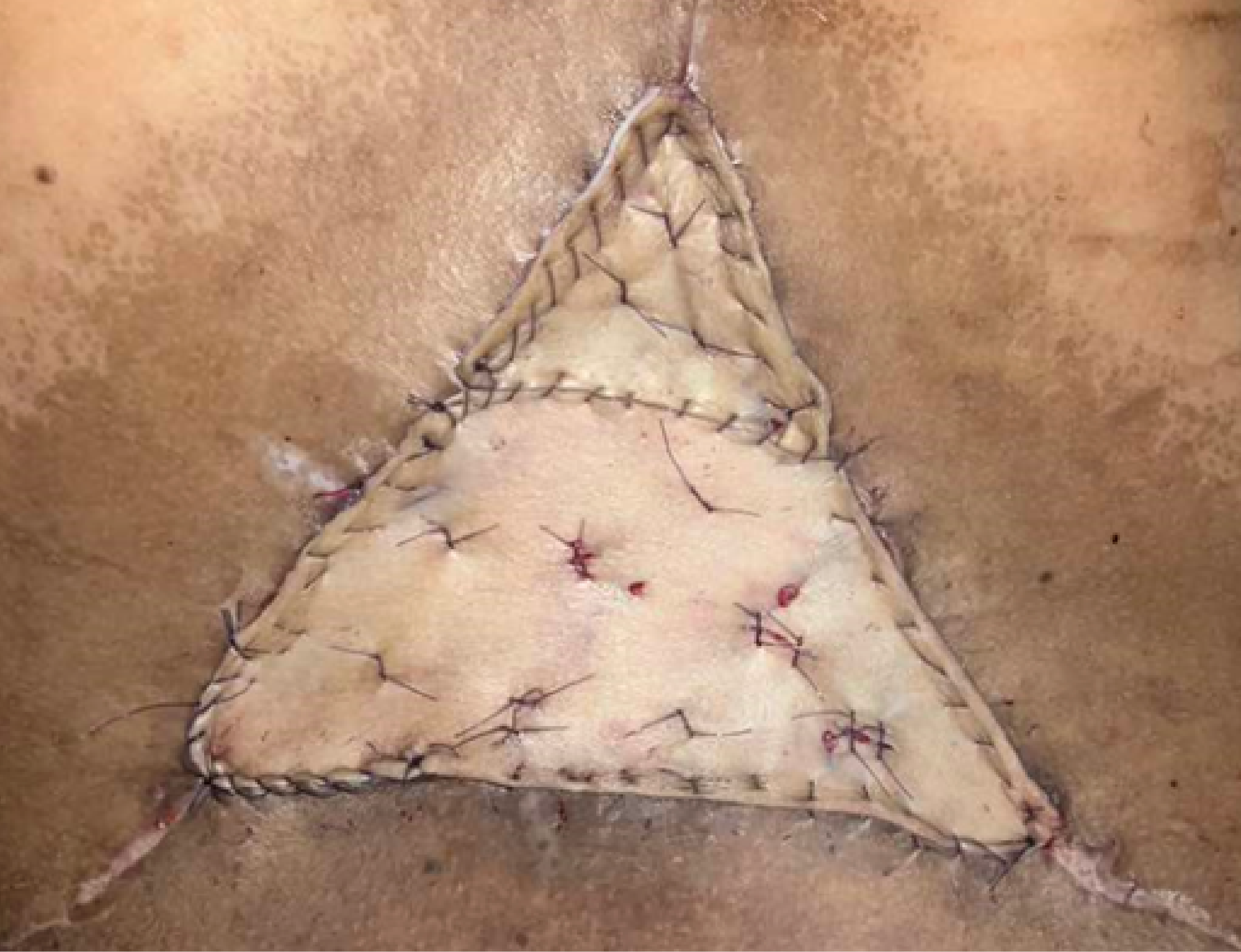
The patient was subsequently readmitted for definitive surgical management and underwent wide tumor resection of the lumbar mass with intraoperative frozen section biopsy. Postoperatively, she developed a surgical site infection complicated by wound dehiscence and febrile episodes. Broad-spectrum intravenous antibiotics, including vancomycin, meropenem, and ciprofloxacin, were initiated. Wound cultures yielded Candida albicans and Acinetobacter baumannii, prompting the addition of intravenous fluconazole and colistin while continuing prior antibiotics. The infection was managed with surgical debridement and two cycles of vacuum-assisted closure therapy, followed by placement of a full-thickness skin graft harvested from the bilateral gluteal folds (Figure 9). After resolution of the infection, the patient was discharged and later readmitted for adjuvant chemotherapy.
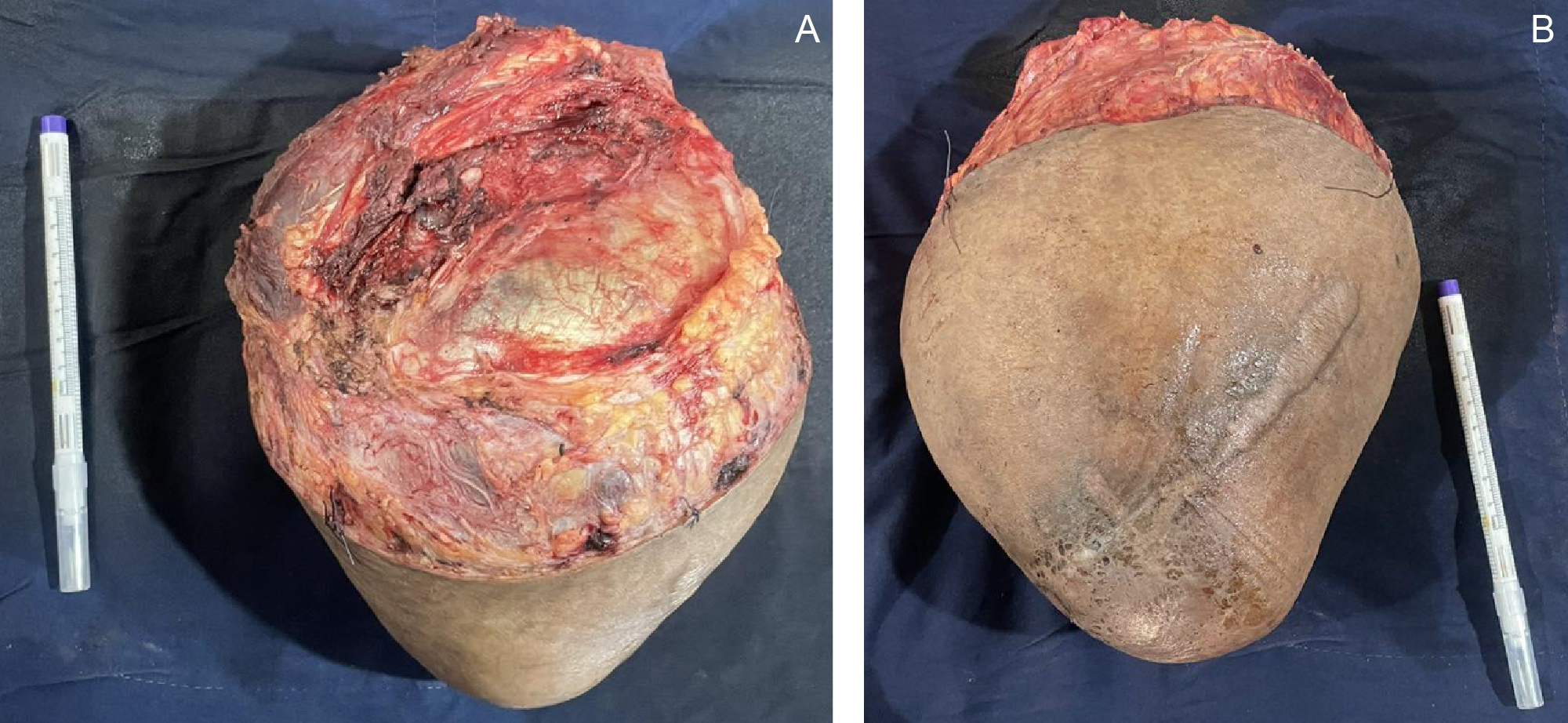
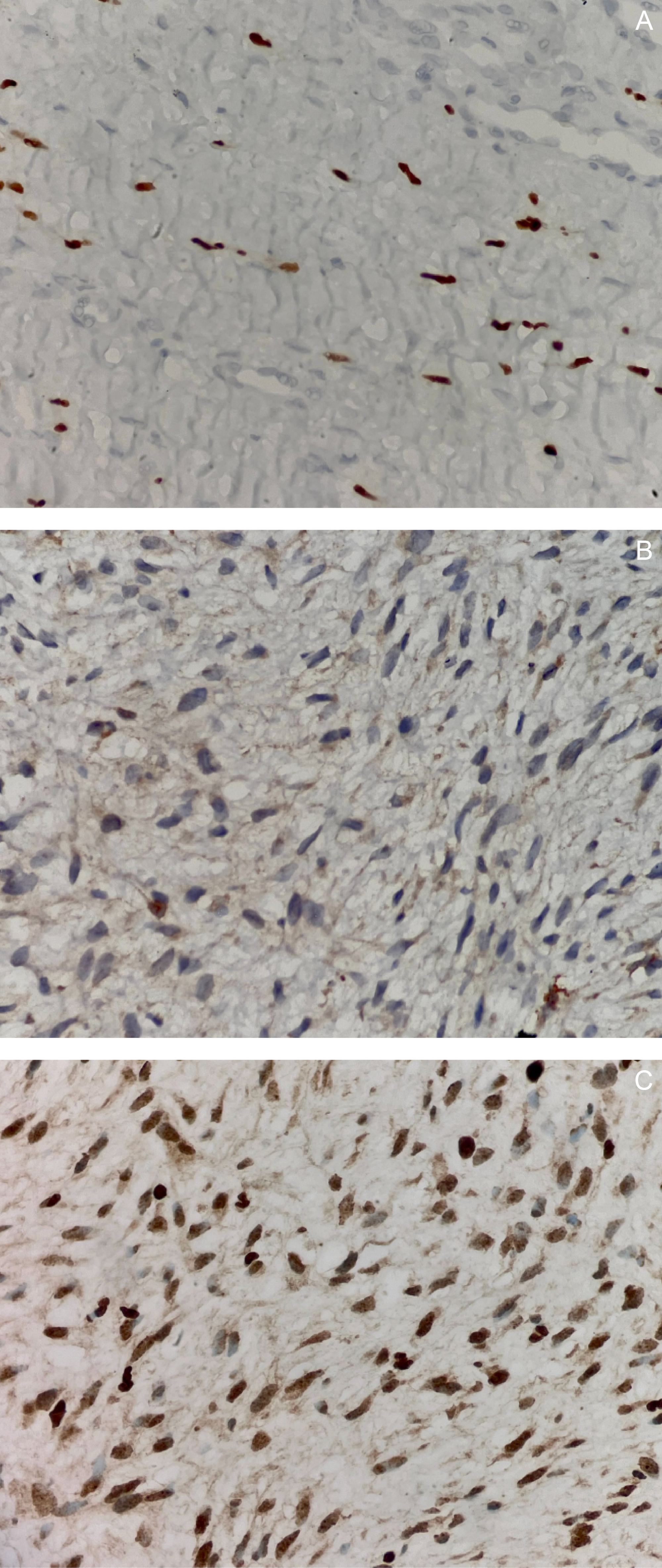
Gross examination of the resected specimen revealed a well-circumscribed mass measuring 16 × 16 × 7.5 cm (Figure 5). Serial sectioning demonstrated approximately 70% necrosis and 40% hemorrhage. Microscopically, the tumor exhibited a marbled low-power appearance with a fascicular growth pattern of atypical spindle cells; all surgical margins were negative for tumor involvement (Figure 6). Additional IHC staining showed focal SOX10 positivity and a mosaic pattern of H3K27me3 expression, indicating partial loss (Figure 7). These histopathologic and immunohistochemical findings were consistent with a low-grade malignant peripheral nerve sheath tumor (MPNST), although the presence of high-grade components in unsampled areas could not be entirely excluded.
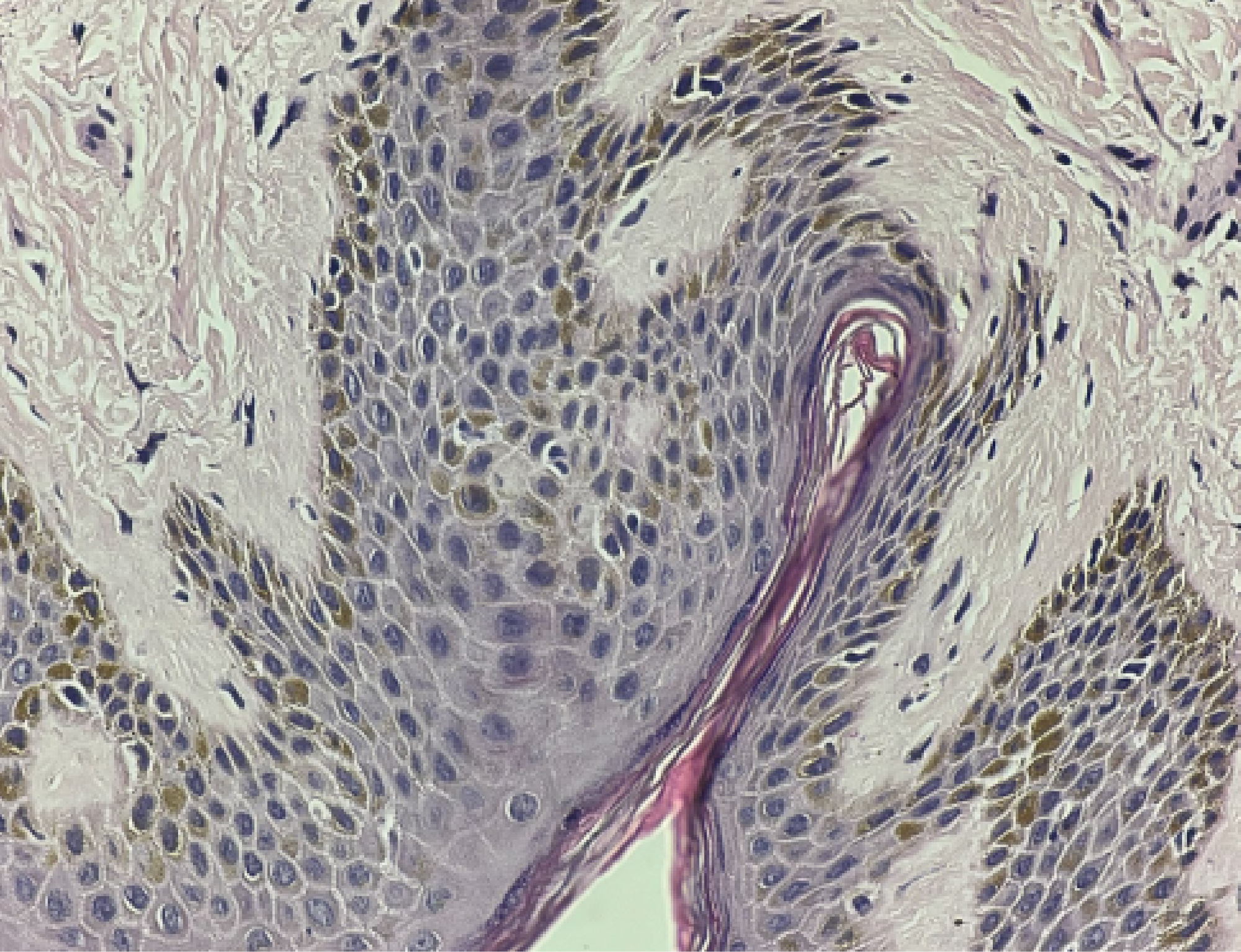
The hyperpigmented patch was further evaluated by Pediatric Dermatology to assess for neurofibromatosis type 1 (NF1). Histologic analysis revealed keratinocyte hyperpigmentation without atypical cells (Figure 8), a finding that may be seen in NF1; however, in the absence of other clinical manifestations, the lesion was classified as a giant congenital melanocytic nevus. Genetic testing was recommended to definitively exclude NF1. The patient was discharged with a final diagnosis of MPNST arising in association with a giant congenital melanocytic nevus. Comprehensive counseling was provided by the Pediatric Oncology Service regarding chemotherapy goals, potential adverse effects, and the risk of recurrence or metastasis. The Orthopedic and Plastic Surgery teams also provided detailed postoperative wound and graft care instructions.
Two weeks following discharge, the patient initiated adjuvant chemotherapy with a doxorubicin–ifosfamide regimen and successfully completed seven cycles. Additionally, she underwent external beam radiation therapy, receiving a total dose of 30 Gy delivered in 30 fractions. Post-radiotherapy CT imaging showed no evidence of recurrent or metastatic disease in the chest, abdomen, or pelvis. At present, the patient remains in good health with no radiologic signs of disease recurrence. She is functionally stable and is preparing to return to school after one year.
Relevance
MPNSTs are rare malignant mesenchymal neoplasms, accounting for approximately 5–10% of all soft tissue sarcomas1 and 3–10% of pediatric sarcomas.2 They affect approximately 0.001% of the general population, with 40% arising sporadically, 50% associated with NF1, and 10% occurring in patients with a history of radiation exposure.3 4 Anatomically, MPNSTs most commonly arise in the soft tissues of the extremities or trunk; therefore, involvement of the lumbar spine is considered an exceedingly rare presentation.5 6 7 Historically, MPNSTs lack distinct clinicopathologic features or defining molecular alterations but are typically associated with loss of function of the NF1 tumor suppressor gene, followed by additional genetic abnormalities such as CDKN2A inactivation and polycomb repressive complex (PRC) protein alterations that contribute to malignant transformation.8 9 10Establishing a definitive diagnosis remains challenging, as clinical presentation varies depending on anatomic location; lumbar spine tumors may manifest with limb pain, abdominal or back pain, urinary or bowel dysfunction, or may be entirely asymptomatic.10 11 Additionally, MRI findings of MPNSTs and benign neurofibromas often overlap, rendering radiologic differentiation difficult.13 14 Consequently, histopathologic evaluation with an extensive immunohistochemical panel is essential to exclude other malignancies.15 16 Although no single immunohistochemical marker is pathognomonic, 50–60% of MPNSTs demonstrate scattered S100 positivity and weak to moderate CD34 expression.15 Complete or mosaic loss of H3K27me3 expression has emerged as a valuable diagnostic marker in confirming MPNST.17
The primary treatment modality is surgical excision with negative margins to minimize recurrence risk and improve survival outcomes.18 In tumors arising at rare anatomic sites, treatment typically consists of surgery with adjuvant therapy. For tumors larger than 5 cm, adjuvant chemotherapy with doxorubicin and ifosfamide is generally recommended.1 19 Despite aggressive multimodal management, five-year local recurrence rates range from 27.3% to 85.7%,18 and overall survival averages 34–52%, corresponding to a median survival of 5–8 years.20 Overall, prognosis for patients with MPNST remains poor due to the tumor’s rarity, aggressive behavior, and high propensity for recurrence and metastasis.1
In our patient, the clinical presentation, radiographic findings, and initial histopathologic evaluation were highly suggestive of spindle cell carcinoma; however, an extensive IHC panel ultimately excluded this diagnosis. The case was particularly challenging due to multiple radiologic, histologic, and immunohistochemical features that mimicked other tumor entities. Furthermore, the absence of NF1 association, lack of prior radiation exposure, and the tumor’s rare anatomic location added to the diagnostic complexity. A definitive diagnosis was established only after comprehensive IHC analysis of the resected specimen. Given the aggressive behavior of MPNSTs, early and accurate diagnosis is critical, as delays may result in more complex surgical management, reduced responsiveness to chemotherapy, and poorer survival outcomes. In this case, imaging and routine histologic evaluation were insufficient, and confirmation required the integration of multiple immunohistochemical markers to reach the correct diagnosis.